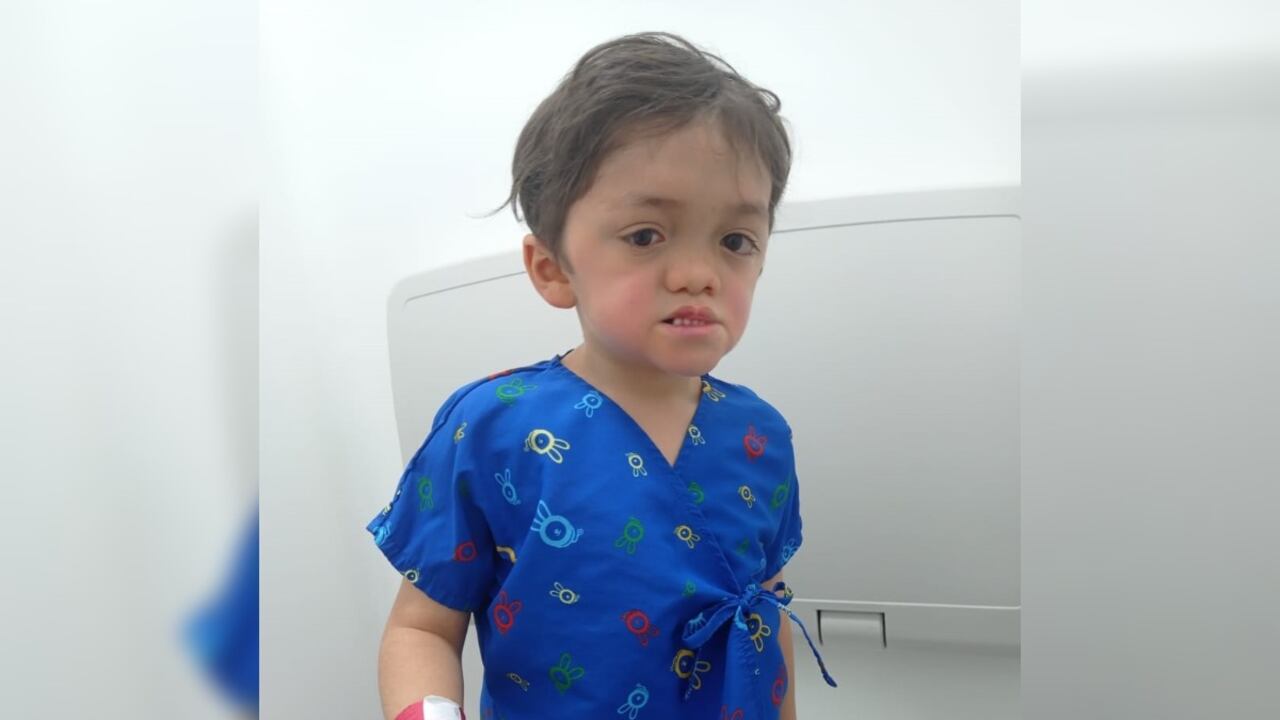
Conocer a Juan David Lizcano Serrato es una oportunidad para remontarse a los mejores años de la niñez y con ello, la inocencia de la vida y el positivismo que todo irá mejor con el pasar del tiempo. Sin embargo, en medio de la ternura que produce cada ocurrencia del menor de 6 años, existe la premura de sus padres por ayudarlo a que su condición médica mejore y con ello pueda disfrutar de su niñez y adolescencia de la mejor manera posible.
Como ya es sabido, en el mundo existen miles de personas que, pese al avance de la ciencia, deben sobrellevar difíciles y complejas afecciones en su salud como consecuencia de las denominadas enfermedades huérfanas o raras, definidas por el Ministerio de Salud como aquellas que afectan a un número pequeño de personas en comparación con la población general y que, por su rareza, plantean cuestiones específicas.
Entre este tipo de enfermedades está el Síndrome de De Morsier, también conocida como displasia septo-óptica, una de estas enfermedades raras que afecta entre otras cosas el hipotálamo, parte del encéfalo encargado de controlar el funcionamiento del sistema nervioso y que por lo tanto, afecta gravemente la salud de quienes la padecen.
Ese es justo el padecimiento de Juan David, que desde que nació viene sufriendo de diversas complicaciones médicas que le afectan su capacidad visual, así como su desarrollo hormonal y cognitivo. La enfermedad, según María Fernanda Serrato y Juan Francisco Lizcano, padres del pequeño y quienes conversaron con SEMANA, le fue diagnosticada a los dos meses de nacer, esto luego de varios exámenes que le fueron practicados al presentar algunas complicaciones a los días de nacido.
“La mayoría de quienes lo tienen presentan solo algunos de los síntomas; JuanDa, en cambio, los tiene todos: nació con labio y paladar blando fisurado, con obstrucciones en la nariz y la garganta que le impedían comer con normalidad, y con una hernia cerebral en la mitad de la cabeza, justo detrás de su nariz. De eso fue operado a los 3 años, y en unos meses pasará por otra intervención”, explicó la madre de Juan David.
Según la mujer, el pequeño JuanDa, como le dicen de cariño sus familiares y amigos, tampoco “tiene cuerpo calloso, ni quiasma, ni un nervio óptico. El otro nervio tiene malformaciones, lo que reduce su capacidad de visión, y su glándula pituitaria está afectada, lo que provoca un déficit de cinco hormonas que nosotros debemos suplir con inyecciones y medicamentos a diario. Sus órganos vitales, de momento, no han estado afectados. Aparte de esas dificultades, tiene un retraso global del desarrollo de entre el 30 y el 40 %. La enfermedad hace que tenga también algunas características de autismo”.
“El bebé nació con malformaciones cerebrales. Yo no lo sabía desde el embarazo, esto no fue diagnosticado desde la gestación, sino al momento de nacer. Él nació con su labio leporino, paladar también algo fisurado, paladar blando (…) todo esto le generó una hernia cerebral que hace que su cerebro prácticamente esté escurrido, está caído, y claro, esto le genera un retraso neurológico”, agregó.

De acuerdo con Serrato, los médicos sospecharon de algún padecimiento más complicado que del labio y paladar hendido, debido a que la abertura o división en el labio superior no era en un lado como es común en esta enfermedad, sino en todo el medio, sin embargo, no diagnosticaron la enfermedad de inmediato.
A los diez días de nacido, el niño salió de la clínica, sin embargo, ya en casa, comenzó a sufrir de vómitos repentinos y no retenía la leche que consumía. Por ese motivo, tuvieron que llevarlo de nuevo al centro clínico, donde permaneció dos meses en constantes exámenes donde poco a poco fueron descubriendo cada una de las afecciones que padecía, entre ellas, una complicación con su glándula pituitaria, encargada de producir hormonas que afectan el crecimiento y las funciones de las otras glándulas del cuerpo, resultados con los que concluyeron que se trataba del Síndrome de De Morsier.
Fue en ese momento cuando los padres de JuanDa sintieron que la vida les cambió por completo, no solo por ver cómo su segundo hijo, de tan solo tres meses de edad, tenía que luchar para poder resistir a tantas complicaciones en su salud, sino por todo lo que esto representaba en su día a día como padres y en su relación como pareja.
Lo primero que le dijeron a María Fernanda fue que debía considerar abandonar por algún tiempo su empleo y dedicarse por completo al cuidado del menor, quien debía empezar a asistir a terapias y citas médicas diarias; motivo por el que la mujer, profesional en Comunicación Social y Periodismo y dedicada laboralmente a proyectos sociales, tuvo que dejar de lado sus metas personales para dedicarse al proyecto más importante de su vida: ayudar a su pequeño hijo a tener una mejor calidad de vida sin importar qué sacrificios implique lograrlo.
Por su parte, Juan Francisco Lizcano, padre del menor, tuvo que asumir la carga financiera de la familia, y con ello, garantizarle el bienestar tanto a Juan David, como a su esposa y a Juan José, hijo menor de la pareja, de cuatro años de edad.
“Es muy difícil ser mamá cuidadora de una persona con discapacidad. Es algo que te consume la vida. Una doctora en la clínica me dijo a mí directamente, y no a mi esposo, que por ser la mamá tendría que dejar de trabajar. Mi esposo, por fortuna, sí lo sigue haciendo (...)”, explicó Serrato señalando que la discapacidad empobrece, “es algo que recrudece la desigualdad”.
Así fue como empezó la búsqueda insaciable de estos padres por encontrar casos similares en el país y en el mundo, y con ello, validar las posibilidades de encontrar un tratamiento que ayude a mejorar algunas de los padecimientos del menor.
Con ayuda de los portales web, la pareja conoció el caso de una niña de Estados Unidos con la misma enfermedad que fue sometida a un tratamiento con células madre llevado a cabo en el Hospital Better Being (BBH por sus siglas en inglés) en Bangkok, Tailandia, y que ha mejorado notablemente su condición, motivo por el que están haciendo todo lo posible para llevar al pequeño JuanDa a este país del Sudeste Asiático y con ello, puedan contribuir a la mejora de su deficiencia visual, así como de la barrera sensorial que padece.
“Las células madre se inyectan por vía intravenosa, punción lumbar y / o la inyección retrobulbar dependiendo de la condición personal de cada paciente. Esta combinación única permite una mejor focalización de las zonas afectadas”, explica en su web Beike Biotechnolgy, empresa de biotecnología china con oficinas centrales en Shenzhen (cerca de Hong Kong) y Taizhou. que según informa, se asoció con el Hospital Better Being (BBH), el primer centro acreditado en Asia para la medicina funcional, con el propósito de ofrecer a los pacientes el tratamiento más extenso disponible para tratar la hipoplasia del nervio óptico (HNO) y Displasia Septo-Óptica (DSO) por medio del trasplante de células madre de Beike.
Bajo ese contexto, esta familia viene liderando a través de Instagram una campaña denominada #3milpesosPorJuanDa, con la que esperan que, las personas que se unan a la misma, puedan aportar desde tres mil pesos y con ello, logren reunir antes de febrero del próximo año, 40.000 dólares, es decir, aproximadamente 150 millones de pesos colombianos, costo del tratamiento más los viáticos del viaje, que, según lo informado por el centro especializado tailandés, dura alrededor de 15 días.
“Desde hace dos años estamos recolectando dinero para completar los 40.000 dólares que cuesta el tratamiento más los gastos en Tailandia. Lo hicimos a través de la plataforma Change.org, donde hemos obtenido más de 55.000 firmas, pero nada de donaciones. Ahora, con el poder de las redes sociales, estamos logrando mejores resultados. Pueden buscar en Instagram la cuenta @juandacrece.contuayuda”, explican los padres del pequeño Juan David sobre los propósitos de la campaña que cuenta con más de 2.000 seguidores en la red social, donde además se informan los canales para hacer los aportes voluntarios.
Con respecto al tiempo en el que esperan reunir todo el dinero, la madre del menor explica que es porque quieren viajar con otros cuatro padres de Latinoamérica que también llevarán a sus hijos en febrero a que se sometan al tratamiento, quienes han señalado que lo ideal sería viajar a la vez varias personas de habla hispana.
A la campaña se han unido algunos influencers, así como artistas que han realizado conciertos virtuales con el fin de que más personas se sumen, no solo con donaciones, teniendo en cuenta que debido a la situación económica del país no es fácil, sino también compartiendo la información, y con ello, personas que sí tengan la manera de hacer su aporte, puedan contribuir para que esta familia logre lo antes posible su propósito.
“La mayoría de personas que no tienen el dinero para donar, nos han ayudado compartiendo la información en sus historias, en sus estados. Muchas personas también han compartido la información con sus círculos sociales o laborales. Todo tipo de ayuda es muy valiosa, para nosotros nada es poco. Cada aporte lo valoramos bastante y quedamos muy agradecidos porque se trata de la salud de nuestro hijo”, indicó María Fernanda agradeciendo además a todas las personas que se han sumado.
“Yo sé que esto toma tiempo, pero hay muchas personas que se lo han tomado como si fuera su propio hijo, y comparten, y replican, lo hacen dos, tres veces por semana, de verdad, muchas gracias”, dijo.
De hecho, este jueves 5 de agosto, 15 artistas que se unieron para ayudar a que más personas conozcan del caso y con ello, se aumenten las donaciones, harán un show de comedia y cuentería a las 6:30 p.m. a través de un Live que será trasnmitido por el pefil de Facebook e Instagram de Juan David (@juandacrececontuayuda), donde además, los asistentes podrán participar de la rifa de dos noches de camping en Villeta, cortesía de una compañía de turismo que se sumó con esta donación para los seguidores del menor.
Para finalizar, los padres de Juan David hicieron un llamado para que su caso sirva para visibilizar las dificultades que padecen las familias que tienen un pariente con discapacidad, más aún si se trata de una enfermedad compleja; y con ello, reciban beneficios por parte del Estado que les permita asegurar a las personas discapacitadas a su cargo, una mejor calidad de vida.
“Queremos visibilizar las dificultades que tenemos los familiares cuidadores en Colombia. Aunque a nosotros en medio de todo nos va bien, hay situaciones mil veces más graves que viven otras personas. Queremos hacer un llamado a las autoridades y al Congreso de la República para que legislen a favor de quienes tenemos a nuestro cuidado a personas con alguna discapacidad. Necesitamos beneficios y facilidades reales, tanto para nuestra vida cotidiana como para conseguir empleos: al ser cuidadores, deberíamos tener prioridad en los cupos de educación y de trabajo. Debería ser un factor de focalización de población vulnerable”, concluyeron.
¿De qué se trata el Síndrome de De Morsier?
De acuerdo a la Organización Mundial de la Salud (OMS), existen entre 5.000 y 7.000 enfermedades raras, la mayoría de origen genético, motivo por el que, en las últimas décadas, se ha prestado considerable atención en todo el mundo a los esfuerzos para estimular la investigación, el desarrollo y la comercialización de tratamientos para este tipo de afecciones.
Sobre el Síndrome de De Morsier, enfermedad también conocida como displasia septo óptica, Julián Ramírez Cheyne, médico cirujano, Máster en Enfermedades Huérfanas y Doctor en Genética médica, en conversación con SEMANA, dio a conocer algunas de las características principales de esta afección.
“Para decir que un paciente tiene displasia septo óptica o Síndrome de De Morsier, uno se fija en que el mismo tenga una triada de manifestaciones. Sin embargo, no todos los pacientes con la enfermedad tienen todo, pueden tener solamente dos de ellas, por ejemplo. Realmente apenas el 30 % de los pacientes tiene la triada”.
Bajo este contexto, el experto explicó que las afecciones que hacen parte de esta triada son, en primer lugar, la displasia del nervio óptico, es decir una anomalía en el nervio óptico en la que este no está bien desarrollado y que por lo tanto puede conllevar a que el paciente sufra de discapacidad visual.
El segundo componente que hace parte de la triada es la alteración de la hipófisis. “La hipófisis es una glándula que está en el sistema nervioso central que recibe estímulos y libera hormonas, y esas hormonas estimulan a otras glándulas y con ello se regulan determinadas funciones del cuerpo. Por ejemplo, la hipófisis libera la TSH, que es la hormona que va a la tiroides y la estimula para que la misma libere sus productos; la hipófisis libera hormonas como las gonadotropinas, que van a las glándulas sexuales -a los ovarios y a los testículos- y hacen que estos liberen sus productos”, motivo por el que los pacientes también podrían padecer múltiples enfermedades derivadas de esta alteración, entre ellas, hipotiroidismo o hipogonadismo, entre otras, explica el doctor Ramírez.
El otro componente de la triada característica del Síndrome de De Morsier lo constituyen los defectos de la línea media cerebral, es decir, “malformaciones en las estructuras que están en la parte medial del cerebro, como la agenesia o anomalía en el cuerpo calloso y la agenesia del septum pellucidum”.
Teniendo en cuenta esto, aunque las consecuencias de esta enfermedad son muy relativas y dependen de cada paciente, estas tres anomalías son las más frecuentes en quienes la padecen.
Por otra parte, es importante decir que “el síndrome de De Morsier además de tener estos tres componentes, puede tener otras anomalías adicionales a estas tres, por ejemplo, el paciente puede tener retraso mental, diabetes, obesidad, sordera, alteraciones del sueño”, explicó el médico a SEMANA.
Con respecto a la detección de la misma, Ramírez Cheyne indicó que teniendo en cuenta las características de la enfermedad, es posible que no se evidencie durante el embarazo, sin embargo en el mejor de los casos, algunos pacientes pueden llegar a presentar algunas manifestaciones en su organismo desde el nacimiento que podrían conllevar a que se estudie tempranamente la posibilidad de padecer displasia septo óptica.
Caso contrario, son aquellos pacientes que físicamente nacen normales y que comienzan a desarrollar síntomas durante la infancia.
Sobre las causas, el experto explicó que la mayoría de estos son esporádicos, es decir se desconoce la causa de la enfermedad, sin embargo, hay un porcentaje pequeño de pacientes en los que se ha encontrado una causa genética.
Teniendo en cuenta que se trata de una enfermedad huérfana, el experto dice que en el país no hay mucha información acerca de la misma, y que además el personal de salud está mal entrenado en la detección y tratamiento de este tipo de afecciones relativamente nuevas en Colombia.
“En Colombia no hay datos, esto de las enfermedades huérfanas es relativamente nuevo en el país. Apenas desde el 2016 es obligatorio que cuando veamos pacientes con el diagnóstico, los notifiquemos a quienes vigilan, pero aparte de que sea obligatorio, realmente para que haya una proyección de datos, se necesita de todo un proceso de educación a nivel de todo el personal de salud. En general, el personal de salud está mal entrenado en la detección y el manejo de las enfermedades huérfanas porque precisamente como estas son de baja prevalencia, nuestros pénsum de educación están enfocados a enfermedades más prevalentes, casi no nos enseñan sobre enfermedades raras. Ese es uno de los grandes problemas sobre enfermedades raras porque hay poco conocimiento del personal de salud y por lo tanto los pacientes se demoran mucho tiempo en ser diagnosticados”, indicó.
Por otra parte, estas enfermedades huérfanas definidas como graves, crónicamente debilitantes y que amenazan la vida, al experto le gusta definirlas como “enfermedades que históricamente han sido despreciadas por los profesionales de la salud – al no saber de ellas porque no se les enseña sobre las mismas; así como por las farmacéuticas, porque al ser supuestamente muy pocos los casos, no resulta económicamente rentable producir medicamentos para esas enfermedades que no le van a poder vender a muchos pacientes; y también, menospreciadas por la sociedad en general, que, conociendo la situación de estos pacientes, no le ha puesto cuidado a la misma”, indicó Ramírez Cheyne.
El experto agregó que, por este motivo, la mayoría de pacientes se demoran años en ser diagnosticados o muchos no llegan a ser diagnosticados, y, asimismo, no encuentran tratamientos de fácil acceso para tratar la enfermedad.
Pese a esto, el especialista en enfermedades huérfanas señaló que esto ha cambiado un poco los últimos años teniendo en cuenta que se ha avanzado en normativa nacional, así como en investigaciones sobre el tema, y se han desarrollado una serie de actividades que están enfocadas a que cada vez se tome más conciencia sobre las enfermedades raras.
